CHARMM basics for people like me
CHARMM is a moleuclar modelling program developed by the scientific group associated with Nobel Laureate Martin Karplus in the MD (Molecular Dynamics) community.
Here are some CHARMM scrpts that I use in my work. Usually one would not need to use CHARMM directly but rather through the online GUI server CHARMM-GUI. It is best used to maintain file-format (psf, pdb) compatibility with the CHARMM36 forcefield.
Some preliminaries- ### File formats - PDB (Protein Data Bank) file : The main function of a PDB file is to store atomic name and coordinates. It can also store additional information such as “temperature factor”, unit cell dimensions and angles, crystallographic/NMR conditions. An example is shown below for the PDB ID: 1JSN which is the Avian hemagglutinin. One can download the PDB file from the “Download files” option. On clicking it gives a dropdown menu of other PDB formats as well but for our purpose we will be using the legacy format.
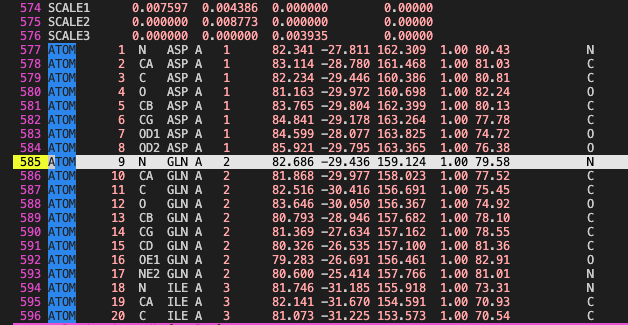
On opening the 1jsn.pdb we see that the first 569 lines are mostly about the experimental conditions. On line 570 we have the CRYST1 line followed by six lines and then by the ATOM lines. For our work we only really the the CRYST1 and the ATOM lines. At the end of the file we have CONECT lines which tell us how the atoms are connected to each other but for us this job is done by the PSF file explained later, so we do not really need it for CHARMM. If you want to manipulate parts of the PDB file it is good to know what each section contains PDB file format. The standard PDB file sometimes runs into issues with the size of the sections. For example, in the ATOM lines only columns 18-20 can contain the residue name. But if the residue name is longer than three characters then it will lead to problems.

CHARMM uses a slightly modified PDB format but this also has similar character length issues which are usually solved by using a different (CRD) file format. In the CHARMM style PDB below obtained from CHARMM-GUI notice the differences-
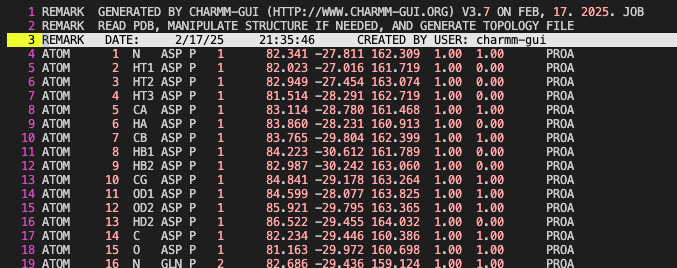
- The hydrogens have been added to the structure which were missing since X-ray, cryo-EM and NMR cannot determine hydrogens many times.
- The atom names have changed to the CHARMM recognized atom types.
- The second last column has changed to identify hydrogen vs non-hydrogen heavy atoms instead of the temperature factors.
- The last column now contains the segment name and not the element name.
CHARMM relies heavily on the residue name (resname), the atom names (HT1, CA…) and sometimes the segname name (segid) to identify the atoms correctly.
For example, the resname ASP above must be there in the CHARMM forcefield to be recognized otherwise the user will have to add it as a new residue. Further, the order of the atoms in the residue must match that of the residue in the forcefield. So in the residue ASP, the first atom must always be the N-terminal nitrogen followed by three hydrogens. Notice also how the three hydrogens have different names HT1, HT2, HT3. The names are just to identity and diferentiate them from each other, however the forcefield contains more information on their specific parameters (I will probably write briefly about the forcefield later).
- CRD (Coordinate) file : Another file format whcih stores the coordinate information but allows for loger names. Confusingly, Amber and CHARMM both use crd as a file extension but have different formats. I am not familiar with the Amber crd format so I will only discuss the CHARMM crd format.It
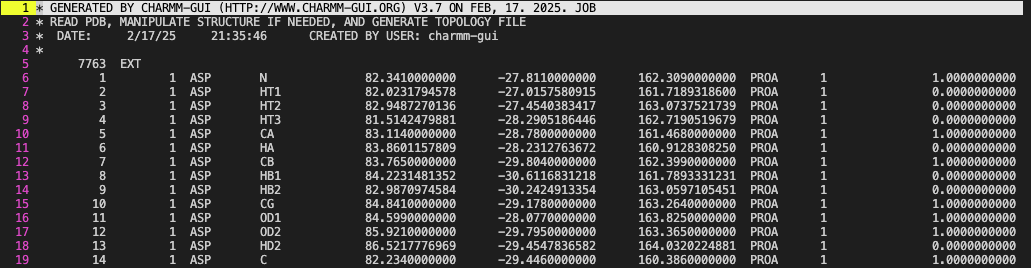 It stores the same information as PDB but the segment name
It stores the same information as PDB but the segment name segid can be used to identify residues instead of the resname. The segid can support longer names.
- PSF (Protein Stucture File) file : This file stores the connection information between atoms. This function is done by the CONECT terms in the standard PDB and they are also used in OpenMM. The PSf file is more convenient and reliable for use with CHARMM.
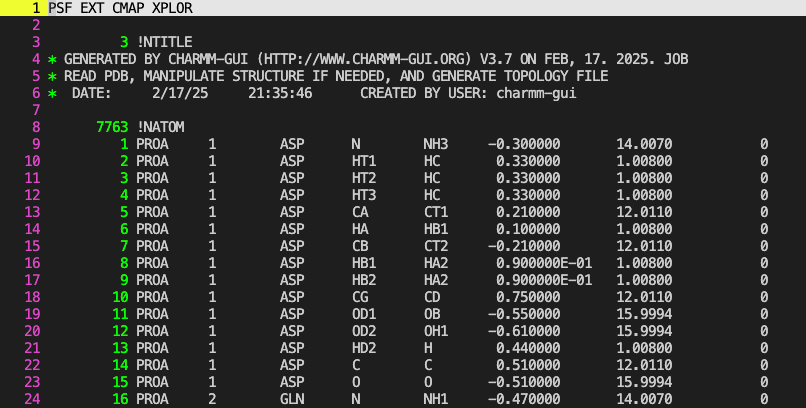 The second column contains the
The second column contains the segid and this must match the one in the CRD/PDB file. The fifth column contains the atom-name (N, HT1, HT2, HT3) same as the PDB and CRD. But this is followed by the sixth column with atom-type (NH3 and HC). So we find that all three hydrogens although have different names have the same type (HC). Thus, they share the same CHARMM parameters (like identical triplets with unique names). The seventh column contains the partial-charge of the atom-type followed by the atomic-mass in the eigth column. The PSF file has more sections below the “NATOM” section - “NBOND”, “NTHETA”, “NPHI”, “NIMPHI”, “NDON”, “NACC”, “NNB”, “NGRP”, “NCRTERM”. All together describe how the atoms are bonded to each other based on the pre-existing residues/molecules in the forcefield.
CHARMM script to read and write
Easiest script I know to read a PDB/CRD file and write the PSF/CRD/CHARMM-PDB files. But this is helpful only for custom, small-molecules not large proteins. For that CHARMM-GUI will be better.
* Read in a PDB/CRD file and ouput PSF
*
DIMENS CHSIZE 2000000
bomlev -3
! Read the charmm ff files
! topparstr contains the path to all charmm ff files
stream toppar.str
! read the sequence in the PDB
read sequence MOL [NMOL] ! Read in the NMOL number of molecule named MOL
generate MOL ! generate the segment with name MOL
! read the sequence of ions in the PDB
read sequence ION [NION] ! Read in the NION number of molecule named ION
generate ION noagle nodihedral ! generate the segment with name ION and don't add angles, dihedrals
read sequence TIP3 [NWAT] ! Read in the NWAT number of water molecues
generate TIP3 noagle nodihedral ! generate the segment TIP3 and don't add angles, dihedrals
! Read in coordinates from CRD
open read unit 30 card name mol.crd
read coor card unit 30 resid
!! If reading coordinates from pdb
! read coor pdb unit 30 resid
close unit 30
! Write the PSF File
open write unit 31 card name mol.psf
write psf card unit 31
close unit 31
! Write the CHARMM-PDB
open write unit 32 card name mol.pdb
write coor pdb unit 32
close unit 32
stop[# TODO] Add explanation of CHARMM-GUI input script